Accelerated Change: Understanding the FDA’s Expedited Pathways
Jamie Gault outlines the key differentiators between each of FDA's programs to facilitate and expedite drug development and review of new drugs.
Patients want access to medicines sooner. A 2012 Quintiles survey of patients living with a chronic disease found that those with the greatest unmet health need are willing to accept more uncertainty about new therapies provided these therapies offer some potential to improve their health. Of those surveyed, 71 percent stated that it takes too long for drugs to be made available forcing patients to go without potential beneficial therapies and putting lives at risk. These results demonstrate a need for drug developers and regulatory agencies to work together to expedite drug development, without compromising patient safety.
Over the years, the FDA has offered programs to facilitate and expedite drug development and review of new drugs. Most recently with 2012’s FDA Safety and Innovation Act (FDASIA), the FDA began implementing more programs to expedite the development and review of innovative new medicines intended to address unmet medical needs and life-threatening diseases or conditions.
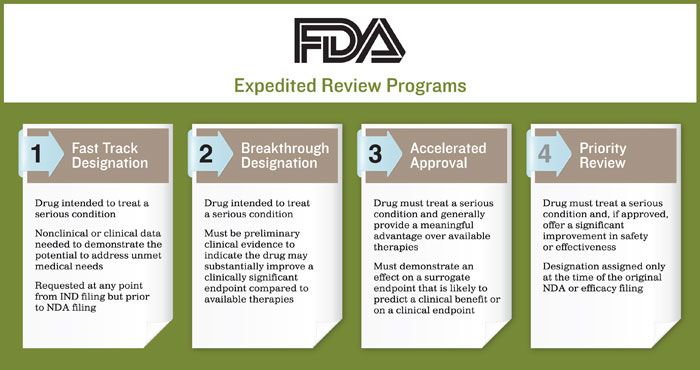
Currently, the FDA has implemented four expedited review programs. The purpose of each is to help ensure therapies for serious conditions are approved and available to patients as soon as it is concluded the benefits justify their risks, while still preserving the standards for safety and efficacy:
- Fast Track Designation
- Breakthrough Designation
- Accelerated Approval
- Priority Review.
To qualify for these expedited programs, the drug must be intended to treat a serious condition - meaning a disease or condition associated with morbidity that has a substantial impact on day-to-day functioning. The drug must also address an unmet medical need such as a condition whose treatment or diagnosis is not addressed adequately by available therapy. It should be noted that rare diseases also qualify for expedited programs. However, because there are development differences and challenges between a rare disease and a common disease, the FDA is more flexible in these situations in order to address the unique challenges.
Drug developers must meet certain criteria to be eligible for a specific program. It’s important to set your overall objective before deciding which pathway will be more beneficial, as each program offers its own specific benefits. In May 2014, the FDA published its Guidance for Industry: Expedited Programs for Serious Conditions - Drugs and Biologics to provide developers with a single point of reference on the different types of programs available as well as criteria to help determine the best option.
Here, we outline the key differentiators between each program and provide some real-world case examples from developers we have worked with to demonstrate how to successfully navigate these pathways to approval.


Fast Track Designation
To qualify for a fast track designation, the drug must be intended to treat a serious condition. Some nonclinical or clinical data must be available to demonstrate the potential to address the unmet medical need. Developers can request a fast track designation at any point from the initial investigational new drug (IND) filing, but prior to the new drug application (NDA) filing.
If no therapy exists for a serious condition, this demonstrates an obvious medical need. The issue comes when there is available therapy for the condition. The development program would then need to demonstrate an unmet medical need still exists such as showing there is:
- an improved effect on the serious outcome of the condition
- an ability to provide benefit in patients who are intolerant to current therapy
- a similar benefit with less serious toxicity that is common and causes discontinuation of treatment.
Many of the benefits of a fast track designation do not apply after a drug developer holds its pre-NDA meeting with the FDA. Submitting a request prior can help ensure an organization receives the full benefits of this program.
Obtaining a fast track designation provides actions to expedite development and review, most notably the rolling submission and review of different sections of the NDA prior to final submission of the clinical data.
Real World Example
In this case a biopharma company developing a drug to treat a serious condition for which there was currently no drug on the market obtained fast track designation at the initial IND filing. This designation was based on the preclinical and clinical data available at the time of the filing. Throughout the course of the trial, the company developer was able to secure frequent meetings with FDA officials, including several informal meetings with the anti-viral division - all of which allowed for accelerated drug development. The company sponsor submitted preclinical and Chemistry, Manufacturing and Controls (CMC) data as part of the rolling review. The NDA is currently under review with the FDA.
Breakthrough Designation
Breakthrough designation is the newest expedited program, established through a 2012 request from Congress. Unlike fast track designation, which is specific to therapies that address an unmet medical need, to be eligible for breakthrough designation, the drug must be intended to treat a serious condition. There must also be preliminary clinical evidence indicating the drug may demonstrate substantial improvement on a clinically significant endpoint over available therapies.
The benefits to breakthrough designation include intensive guidance on efficient drug development and commitment from the FDA to work together to expedite development. Developers are also permitted to submit via the same rolling review process as the fast track designation.
As discussed at the FDA/CMC Summit in December 2014, the FDA’s two year assessment of break through designations has shown that clinical development is often not the rate limiting step in approval. Often the manufacturing development and scale up of the drug lag behind and must be accelerated to allow for a quicker approval. Although the number of requests and designations has exceeded expectations (30% of requests granted), there are still a higher number of denials (49%), as reported by the FDA. Reasons for denial include:
- the evidence provided does not contain clinical data
- clinical data is too preliminary to be considered reliable
- failure to demonstrate a substantial improvement over available therapies.
The FDA is clear about what needs to be included in the request and companies must adhere to those requirements.
Real World Example
Another company developing a drug to treat a rare disease, for which no approved therapy exists, was granted both a breakthrough therapy designation and orphan drug designation. The breakthrough therapy designation was awarded after the company’s initial IND filing and was based on limited data from an investigator-initiated trial.
To date, the FDA has committed to working closely with the company to expedite development of the drug including granting multiple formal meetings --including both Type B and Type A -- within the last year. In addition, the FDA has been working collaboratively with the company helping to determine how much data would be needed to support its marketing application for a rare, orphan indication and recommending a rolling review so the preclinical and CMC data could be submitted while the Phase III clinical trial remained underway.
Accelerated Approval
For the FDA to grant a drug developer access to the accelerated approval pathway, the drug must treat a serious condition and generally provide a meaningful advantage over available therapies. It must also demonstrate an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit or on a clinical endpoint. The effect needs to be measured earlier than irreversible morbidity (IMM) - or mortality that is reasonably likely to predict an effect on IMM or other clinical benefit.
Accelerated approval allows developers to market the drug while it continues conducting a confirmatory study to obtain full approval. This is because approval is based upon an effect on a surrogate endpoint or an intermediate clinical endpoint that is reasonably likely to predict a drug’s clinical benefit.
Real World Example
A company was developing a new drug for hormone refractory metastatic prostate cancer. The only available treatment option was with a chemotherapy (docetaxel)-containing treatment regimen. The developer received accelerated approval within 78 days from the date of the official submission. Communication and collaboration with FDA was key to this approval. Although the company did not have fast track designation, the FDA allowed the sponsor to submit the preclinical and CMC information while the Phase 3 study was ongoing. They even reviewed and provided CMC comments prior to the clinical data ever being submitted, thus demonstrating the power of the rolling review.
The NDA was submitted to the FDA in electronic common technical document (eCTD) format. Though not required, the company met with the FDA for a “navigation meeting,” walking through the electronic NDA so that they could better and more efficiently review the application.
During the review, the company received daily requests from the FDA. It was mutually agreed that rapid email responses would suffice, followed by official amendments to the NDA. This agreement also allowed for batching of the amendments, which in turn helped to further accelerate the process.
Priority Review
For an application to be given priority review, the drug must treat a serious condition and, if approved, offer a significant improvement in safety or effectiveness. This designation is assigned only at the time of the original NDA or efficacy filing and not before, as is seen with fast track or breakthrough designation.
Priority review guarantees a shorter review cycle as the marketing application must be reviewed within six months as compared to a standard review, which can take as long as 10 months.
Real World Example
In 2014, a sponsor received priority review for their once daily, single-entity hydrocodone bitartrate tablet. The company reformulated an already approved tablet to incorporate abuse-deterrent properties designed to make the product more difficult to manipulate for the purpose of misuse or abuse by various routes of administration (e.g. chewing, snorting or IV). This change in formulation offered a significant improvement in safety. Consequently, the company was granted a priority review, allowing for a quicker approval.
In response to the need to hasten drug development and regulatory approvals, the FDA has provided developers with new tools to help expedite development programs and speed access to new products. By understanding these new programs and working closely with the FDA to determine which pathways will best support your goal, we can move potential forward. In the words of Henry Ford, “Coming together is a beginning, keeping together is progress, and working together is success.”
References
- FDA Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics; May 2014
- Quintiles: The New Health 2012 Report: Rethinking the Risk Equation in Biopharmaceutical Medicine
- Orphan Drugs Around the World: The Regulations and Requirements You Need to Know; www.raps.org
About the Author
Jamie Gault, RAC, leads Novella Clinical's global regulatory affairs department.

















Navigating Distrust: Pharma in the Age of Social Media
February 18th 2025Ian Baer, Founder and CEO of Sooth, discusses how the growing distrust in social media will impact industry marketing strategies and the relationships between pharmaceutical companies and the patients they aim to serve. He also explains dark social, how to combat misinformation, closing the trust gap, and more.


FDA Grants Priority Review to Regeneron’s Eylea for Macular Edema Following Retinal Vein Occlusion
April 18th 2025Regulatory action was based on data from the Phase III QUASAR trial, which demonstrated that Eylea HD dosed every eight weeks achieved non-inferior visual acuity outcomes compared to Eylea in patients with macular edema following retinal vein occlusion.

