Clinical Trials Don't Have to Cost Too Much or Take Too Long
Pharmaceutical Executive
A discussion of the cost and time put into clinical trials.
The pharmaceutical research landscape is littered with the remains of failed clinical trials. Since 2008, 17.2% of Phase 2 trials and 12.2% of Phase 3 trials have been prematurely terminated, according to an analysis of the Phesi database, which comprises more than 320,000 clinical trials and over 500,000 investigators across several hundred disease indications. Given that estimated global pharmaceutical R&D spending currently amounts to $125-$160 billion annually,1,2 those terminations mean roughly $20 billion of that spending is essentially wasted every year. More importantly, terminated trials dash the hopes of patients who could have potentially benefitted from the medical innovations that might have emerged from successful trials.
The Phesi database reveals that patient recruitment difficulties are responsible for 57% of failed Phase 2 trials and 54% of failed Phase 3 trials. Such difficulties result from a variety of factors including suboptimal protocol design, inefficient business processes (especially with regard to site activation), and poor investigator site performance. These difficulties are avoidable and can be addressed through better understanding of the operational characteristics of clinical trials, which itself can lead to improved clinical trial planning.
The perils of inadequate planning
At the risk of oversimplification, a clinical trial collects and analyzes safety and/or efficacy data from a well-defined group of patients in a highly regulated and carefully controlled setting. Depending on how one defines a variable, it may take several dozens or even hundreds of variables to determine the outcomes of a clinical trial. However, even when a trial sponsor (or the CRO it works with) does a hundred things right, one mistake can jeopardize a trial’s success.
Oftentimes, success may hinge on the trial planner’s appreciation of the complexity of the disease, or on a team’s ability to determine the appropriate number of patients, the right number of investigator sites, and the optimal duration of the trial. While each of these factors is a major driver of clinical trial costs, the numbers of patients and sites typically generate relatively little discussion from a financial perspective. Moreover, the clinical trial process is idiosyncratic, dependent on variable experience, and usually conducted without regard to the broader experience of similar trials that have already taken place.
To a great extent, the inattention given to these factors stems from simplistic, perhaps wishful planning and unrealistic, uncalibrated expectations: pharmaceutical companies generally want to get their new medicines to patients as soon as possible, at the lowest possible cost.
The desire for speed can encourage a risky form of linear thinking: for many a Phase 3 trial, the operational model is derived from a successful Phase 2 trial, from which the number of investigator sites is extrapolated in order to attain a similar enrollment cycle time (ECT), which is the elapsed time from first to last enrolled patient, as shown in the following hypothetical example of a clinical program for an investigational anticancer agent:
Phase 2 -- actual
Phase 3 -- planned
Phase 3 -- actual
Patients
160
970
970
Sites
48
280
258
Enrollment cycle time (ECT) (months)
14
12
24
Gross site enrollment rate (GSER) (patients/site/month)
0.29
0.29
0.15
Site effectiveness index (SEI)
0.68
unknown
0.71
Table 1. Planned versus actual patient enrollment metrics for a Phase 3 oncology clinical trial
In the above example, the trial planners used an assumed linear relationship between number of patients, number of sites, and ECT to extrapolate the Phase 2 ECT of 14 months to a forecasted ECT of 12 months for the Phase 3 trial. Unfortunately, such a linear relationship does not exist: the actual Phase 3 ECT was 24 months-twice the forecast. A Phase 3 trial is not just a bigger Phase 2 trial; oftentimes, this is a costly lesson.
The power of predictive analytics
Phesi has developed a predictive analytics platform that consolidates comparable trial and site metrics to support trial design, protocol design, site selection, and trial execution. Although no two trials are exactly alike, the platform yields a mathematical relationship that enables a “comparison of the incomparables,” using the following metrics:3-5
- Site Activation Curve: the number of sites activated over time
- Site Effectiveness Index (SEI): the area under the site activation curve-a relative measure of the available site capacity that is being utilized over time
- The SEI can be measured as the percentage of selected sites open for enrollment over the duration of the enrollment cycle time, or as the percentage of time a single site is open for enrollment compared to the overall trial enrollment duration. As a percentage, SEI is always larger than zero and less than one.4
- Enrollment Curve: the number of patients enrolled over time
- Gross Site Enrollment Rate (GSER): the effective trial-level enrollment rate, expressed as patients/site/month
- Adjusted Site Enrollment Rate (ASER): the product of SEI multiplied by GSER (SEI x GSER = ASER)
- Enrollment Cycle Time (ECT): the elapsed time (in months) from first enrolled patient to last enrolled patient
Using these metrics, one can reliably analyze data from comparable trials (actuals) and a client’s trial (forecast) to reveal patterns behind the numbers. For an extensively studied disease indication, we select a set of randomized clinical trials that are similar to the client’s planned trial in terms of number of patients, number of investigator sites, inclusion/exclusion criteria, and other relevant parameters. We then use those parameters to develop a bubble chart that incorporates three variables: number of activated sites (N), GSER, and ECT, where each bubble represents one selected clinical trial. The size of each bubble reflects the length of ECT, with larger bubbles representing a longer ECT.
A sample bubble chart appears in Figure 1, which shows how adding sites to a trial can suppress individual site performance:


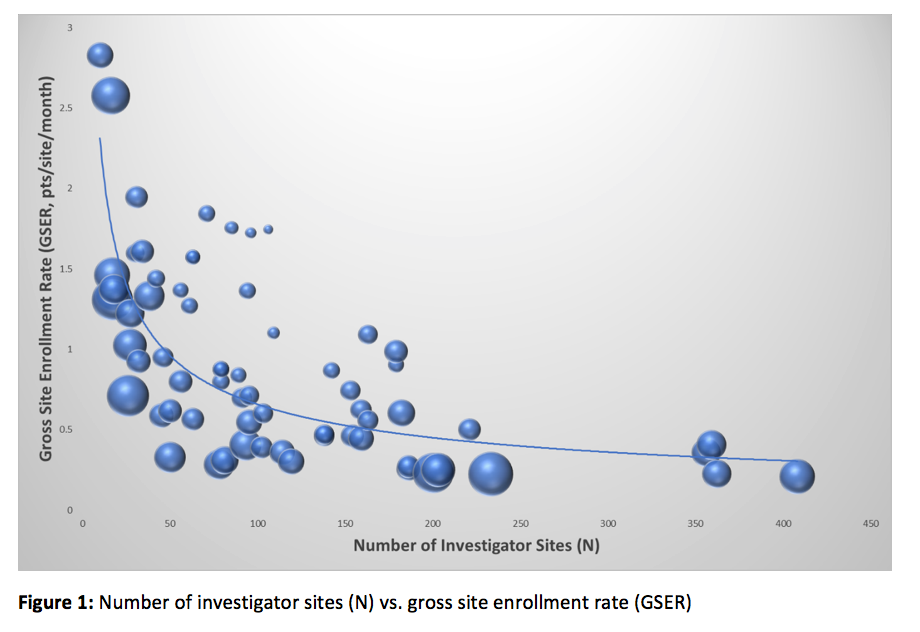
Figure 1: Number of investigator sites (N) vs. gross site enrollment rate (GSER)
It seems intuitive to add sites to a trial in order to have them contribute more patients and thereby reduce ECT. What is less intuitive, however, is that the incremental benefit vanishes at a certain point, beyond which the ECT is prolonged. As Figure 1 illustrates, the declining GSER means each site contributes fewer patients over a defined period of time (ECT). In other words, the point of diminishing returns is reached early in the course of the trial, in part because of slow site activation (a particularly thorny problem for large studies with many sites), in part because the best sites are recruited first. Late activation of a poorly performing site pulls down the site activation curve. This distinctive pattern holds true for over 1,000 different disease indications we have analyzed, and we suspect it is nearly universal.
Figure 2 further pinpoints the optimized scenario at the point where activating 79 sites would yield an ECT of 273 days. Beyond this boundary, the benefits diminish:



As shown in Figure 2, the enrollment and site activation patterns, coupled with the observed mathematical relationships, essentially enable us to objectively determine the optimal number of sites. Moreover, the predictive analytics platform facilitates clinical trial design optimization and country-to-country comparison of site performance, among many other possibilities.
Too many sites
One might argue that even if the GSER decreased, there would still be a surplus of eligible patients to potentially reduce the ECT. But that is not what we get in reality (see Figure 3):


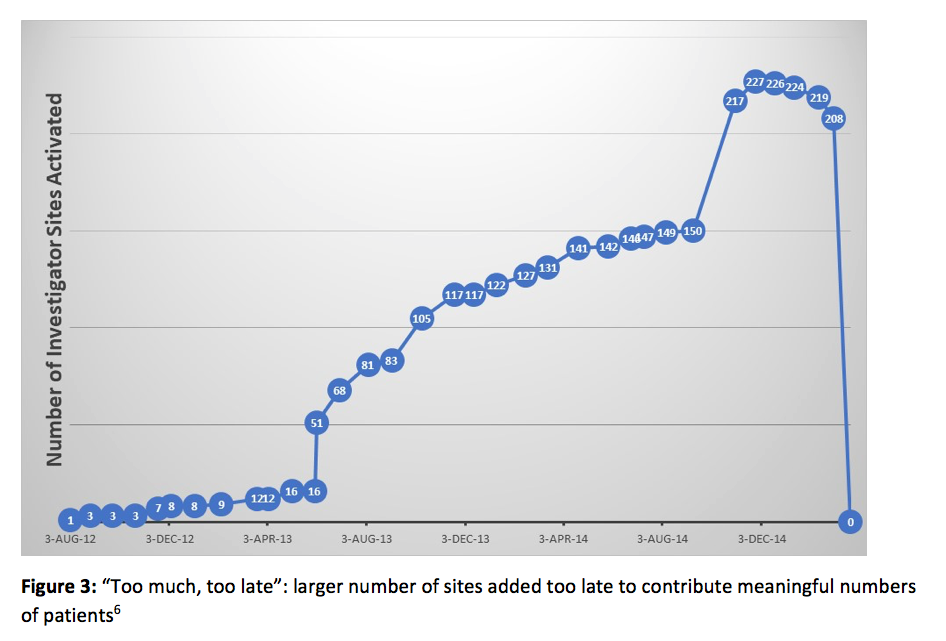
Why do the benefits fall off so dramatically? It’s because activating an excessive number of investigator sites yields a larger trunk of non-performing sites that drain financial resources and, in all likelihood, prolong the ECT. In the example illustrated in Figure 3, a total of 227 sites were activated in this trial, but only about 140 sites contributed patients. Moreover, the 77 sites activated in the last six months of the trial did not contribute a meaningful number of patients. The number of activated sites far exceeded the 120 sites recommended via our optimization analysis, as illustrated in Figures 1 and 2. Additionally, the 87 non-performing sites created a financial exposure amounting to $10.4 million, based on an assumed $30,000 in site activation costs and $3,000 per site per month over a 30-month duration. Those costs yielded an SEI of 44%, significantly lower than the recommended 60% SEI value for this trial.6
The disparity between actual and recommended SEI illustrates one of the perils of activating too many investigator sites: activating a large number of sites takes time, especially in the early stages of a trial. In the trial described above, the team was forced to push too many sites forward with limited resources, and a large percentage of sites were activated near the end of the ECT, when the team was spread too thin by focusing on too many nonproductive tasks, and was unable to focus on maximizing returns from the most productive sites. In short, there is such a thing as “too big to succeed.”
Too few sites
Moving in the opposite direction risks crossing another boundary, one that results from activating too few sites rather than too many. Such a situation may occur when a budget-conscious sponsor funds an insufficient number of sites (see Figure 4):


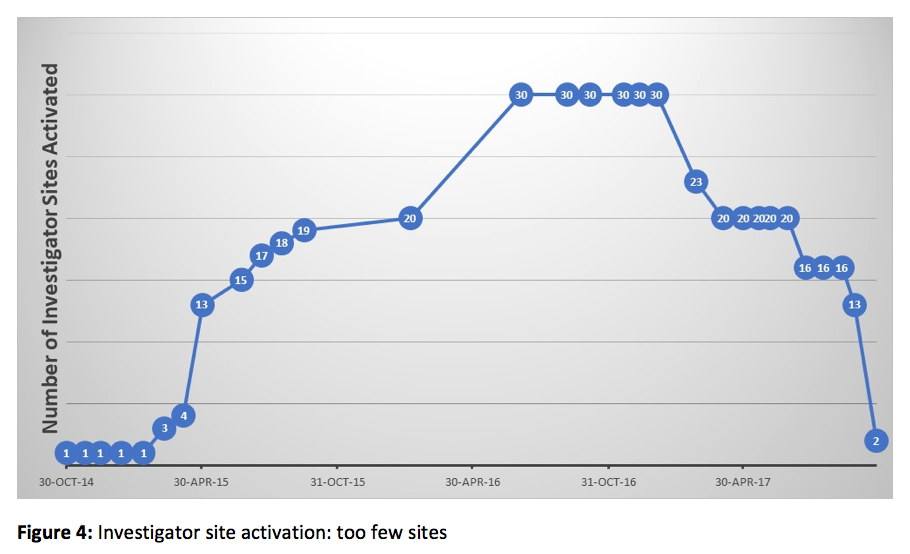
In this case, analysis of our database yielded a recommendation of 80 sites and a forecasted ECT of 15 months. The trial team, restricted by available funding, decided to activate 30 sites instead. The lower number of sites reduced site activation costs by about $1.5 million. The trial team used these savings to extend site management over a much longer time frame, from 15 months to 35 months. Unfortunately, the savings were negated by extra costs for drug supply, medical monitoring, and various other project management costs. The 20 extra months in ECT therefore constituted wasted time and a lost opportunity to optimize the site activation timeline. Presumably, advance knowledge of these opportunity costs would have prompted management to make a different decision about this trial.
Enhancing organizational awareness of the boundary
The beauty of the analytics platform is that it is objective and quantitative, enabling trial planning and execution in an integrated fashion. Nevertheless, true integration is not a given. In many big pharma companies, and even in some small ones, siloed decision-makers can jeopardize clinical trial success. Even if the trial planner is aware of the point of diminishing returns (and of the risks of disregarding this critical juncture), this knowledge is irrelevant unless it is shared across the organization. That speaks to the importance of cross-functional communication between the medical, clinical, commercial, regulatory, and finance teams-as well as between sponsor and CRO-to optimize decision-making. When each of these parties understands the importance of the factors that affect site activation and patient enrollment, and of the variables that determine enrollment rates and site performance, the organization as a whole (and its CRO partner) can successfully navigate what might otherwise be a perilous clinical trial landscape.
Gen Li, PhD, MBA, is the corresponding author. He is founder and president of Phesi. He can be reached at gen.li@phesi.com
Stephen Arlington, PhD
Paul Chew, MD
Annalisa Jenkins, MBBS, FRCP
References
- ICON plc presentation at 35th Annual J.P. Morgan Healthcare Conference, San Francisco, Calif., 2017 Jan 10. http://files.shareholder.com/downloads/ICLR/3717560502x0x923636/3ECDE4CD-8149-47D3-86B0-F81B69616D25/ICON_JPM_2017_10th_Jan_Final.pdf.
- Catalent, Inc. presentation at 34th Annual J.P. Morgan Healthcare Conference, San Francisco, Calif., 2016 Jan 11. http://investor.catalent.com/sites/catalent.investorhq.businesswire.com/files/event/additional/2016.01.08_-_CTLT_-_JPM_HC_Conference_FINAL.pdf.
- Li G. Site activation: the key to more efficient clinical trials. Pharmaceutical Executive. 2008 Dec 12. http://www.pharmexec.com/site-activation-key-more-efficient-clinical-trials.
- Li G, Harper B. Finding the sweet spot. Pharmaceutical Executive. 2009 Oct 2. http://www.pharmexec.com/finding-sweet-spot.
- Li G. Forecast enrollment rate in clinical trials. Applied Clinical Trials. 2015; 35(3):42-48. http://www.appliedclinicaltrialsonline.com/forecast-enrollment-rate-clinical-trials-0.
- Legagneur V, Peachey J, Correa K, Li G. Enrollment cycle times can and should be optimized. Applied Clinical Trials. 2018 Jan 17. http://www.appliedclinicaltrialsonline.com/enrollment-cycle-times-can-and-should-be-optimized.











FDA Grants Priority Review to Regeneron’s Eylea for Macular Edema Following Retinal Vein Occlusion
April 18th 2025Regulatory action was based on data from the Phase III QUASAR trial, which demonstrated that Eylea HD dosed every eight weeks achieved non-inferior visual acuity outcomes compared to Eylea in patients with macular edema following retinal vein occlusion.

Addressing Disparities in Psoriasis Trials: Takeda's Strategies for Inclusivity in Clinical Research
April 14th 2025LaShell Robinson, Head of Global Feasibility and Trial Equity at Takeda, speaks about the company's strategies to engage patients in underrepresented populations in its phase III psoriasis trials.

New Insights Into T Cell Exhaustion and Inflammation in Long COVID
April 17th 2025Nigel McCracken, chief operating officer, Virax Biolabs, discusses new findings that reveal altered cytokine activity and evidence of T cell exhaustion in long COVID patients, providing deeper insight into post-infection immune disruption.

Key Findings of the NIAGARA and HIMALAYA Trials
November 8th 2024In this episode of the Pharmaceutical Executive podcast, Shubh Goel, head of immuno-oncology, gastrointestinal tumors, US oncology business unit, AstraZeneca, discusses the findings of the NIAGARA trial in bladder cancer and the significance of the five-year overall survival data from the HIMALAYA trial, particularly the long-term efficacy of the STRIDE regimen for unresectable liver cancer.

Amgen’s Imdelltra Demonstrates Significant Overall Survival Improvement in Small Cell Lung Cancer
April 16th 2025In the Phase III DeLLphi-304 trial, patients with small cell lung cancer administered Imdelltra achieved a statistically significant and clinically meaningful improvement in overall survival compared to standard-of-care chemotherapy.


