A Clinical Sea Change
This prediction model will allow developers to recoup costs and enjoy a longer life cycle under patent protection.
The past few years marked a low point in worldwide market launches for new active compounds. According to the IMS LifeCycle New Product Focus, pharma launched 31 new active substances worldwide in 2004, which was only one more than the 25-year-low in 2003, and down from 37 in 2001. It's not that companies weren't performing R&D—paradoxically, investigational drug applications to FDA reached 495 in 2004, passing a record of 415 that was set in 1998.
The paradox is explained by attrition. According to the business intelligence company Pharma Projects, nearly 750 candidates were in Phase I trials in 2003 (up 85 percent from a decade earlier); nearly 1,250 were believed to be in Phase II (up 90 percent). But the number of candidates in Phase III was about 400, unchanged since the mid-1990s. The cumulative clinical-stage failure rate is now 80 percent, with 30 percent of new molecular entities failing in Phase I (mostly because of inappropriate pharmacokinetic or pharmacodynamic characteristics).
Industry observers contend that outdated study models have been a real obstacle to change in pharma. But as new computational tools and microdosing methods emerge, researchers can help companies change how they conduct studies, to make them cheaper, faster, and more effective. Why the steep failure rates and rising costs? A recent survey by Cambridge Healthtech Associates (CHA) locates specific reasons for the delays and failures of clinical trials in the many deficiencies in current clinical-trials protocols.
Failure to Launch The main reason for delays? Recruitment of patients, mentioned by 68 percent of survey respondents. Just as commonly cited was study initiation, or the process of securing IRB approvals, inking contracts, and developing budgets. The most frequently cited reasons for the termination or failure of clinical projects were problems with efficacy (64 percent), inconclusive trial results (57 percent), and faulty trial design (43 percent). The respondents ranked the top-three reasons for termination of trials in order of priority, and responded as follows: inconclusive trial results, problems with efficacy, and commercial reasons (e.g., compound is misaligned with market need).
Calculating the Spend Pharma is spending more than ever on drug development. While the limited set of preclinical safety studies required to initiate Phase I might cost less than $2 million, the fully calculated cost of conducting a Phase III program can amount to several hundred million dollars easily—and it's rising. An economic model developed in 2003 by Bain Consulting showed that the total investment required to launch a drug increased from $1.1 billion between 1995 and 2000, to $1.7 billion between 2000 and 2002. During this period, the cost of discovery research remained static; the 55-percent increase was due almost exclusively to the clinical stages of drug development.
Passing the Test The real problem emerges when companies become burdened by the drug's post-marketing safety problems, even after it passes through clinical testing. According to a study from the Tufts Center for the Study of Drug Development, 3.5 percent of all the drugs approved for sale in the United States in the 1990s were withdrawn from the market subsequently; the rate has dropped to 1.6 percent since then, but there are numerous cases when approved drugs had to be withdrawn from the market soon after launch because of unexpected adverse effects. This indicates that the present drug-development process does not ensure safety early in clinical development.
While cost doesn't seem to be the issue, the most obvious and urgent objective for the industry is to bring the clinical process under better control to ensure a drug candidate's ultimate success. But the industry's response to increasing clinical attrition has so far consisted mostly of efforts to streamline the existing processes without changing any of the fundamentals.
This conservatism has a good reason, which is unrelated to science and technology: Major deviations from established regulatory pathways to approval would involve the risk of long delays and the probability of regulatory setbacks. Companies tend to move cautiously, mostly by using information technology to make patient data entry easier and less error-prone, but without fundamentally changing the clinical process. CHA's survey found that 45 percent of respondents described the culture or their clinical organization as risk averse.
FDA's Clinical Trials Initiative
Pharma companies might be risk averse, but the industry has seen an increase in more fundamental attempts at change, such as introducing information-rich trial designs. The integration of pharmacogenomics was a relatively quick and easy process, but this has not been the case with pivotal or outcome measures like biomarkers. In its Critical Path document, FDA has outlined possible changes intended to speed the clinical trials process: The agency has signalled that it is willing not only to listen but to take the lead in an interactive process of change that should restore productivity to drug development. The Exploratory Investigational New Drug (IND) Guideline of January 2006 is the first concrete step towards opening the rigid regulatory framework to accommodate more relaxed and pragmatic approaches in Phase I.
Companies are making efforts to implement key clinical technologies and practices into their existing systems (See "Facing Their Future"). There is a relatively high level of investment in biomarkers, EDC, and offshoring opportunities, but the current implementation of microdosing and adaptive design is extremely low. Microdosing allows an investigational drug to be administered to healthy volunteers at doses that are one percent or less of what is expected to have a pharmacological effect, and that must not exceed 0.1 mg. This practice allows an early assessment of pharmacokinetics, and might become a valuable tool for choosing the best of several related candidate compounds for actual clinical development. The rapid ramp-up in investment for microdosing over the next four years is important because it's an example of a restructured approach to clinical drug development—and an example of evolutionary new thinking.


Filling the Gaps
The full integration of information technology is the second arm of the new clinical-trials strategy, with a key role being played by pervasive computing—the trend toward increasingly ubiquitous computing devices in the trial environment. This starts with trial-design modeling, where companies like Pharsight have developed software packages based on existing toxicity information and pharmacokinetic and pharmacodynamic modeling to characterize and minimize the risk of failures. In this "virtual trial space," researchers can quickly explore multiple scenarios to optimize the design of individual trials, increasing the predictability of the process and the likelihood that each trial will give definitive results and lead to a successful program.
A small part of these integrated efforts will be moving from paper-based trial documentation to electronic case report forms (e-CRFs). In the "e-clinical world," the investigator will see the patient's visit information, his study diary information and electronic laboratory results, even the genetic information of the patient. Patient compliance will be monitored by RFID chips, which also record details on how clinical supplies are distributed and stored.
Applied systems biology (the modeling of key features of human organs or the entire human organism relative to health, disease, and pharmacotherapy) and the Virtual Patient (a computational model of human physiology in a particular disease state) are examples of holistic approaches to drug development that are also computation-intensive. Although this technology is still far from validation, partial models, such as Entelos' PhysioLab platform, already have been astoundingly successful. In another line, metabolomic "fingerprinting" of reactions to drugs, which is being pioneered by companies such as Metabolon, will supply clinicians and drug developers with nearly real-time information about metabolic shifts in individual trial participants.
However, technology alone will not solve the problems of clinical and post-launch attrition. To accomplish this, the long-entrenched and rigid structure of the clinical trial process, with its stages I through III, will need to be entirely re-thought (See "Filling the Gaps"). Today's mega-trials, which strain logistic and clinical capacities, will have to be replaced by heavily IT-supported, post-marketing surveillance studies in the real-world environment.


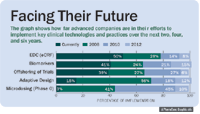
Facing Their Future
The Clinical Trial Process in 2015
In 2015, what is now known as Phase I will assume a new role as a brief confirmatory testing stage of the model for drug and human interactions. Phases II and III will merge into a single, advanced-stage human testing phase that involves fewer patients than today, relying on relatively small populations that are highly homogeneous. Systematic post-marketing studies and an improved surveillance system will be integrated into a monitoring phase that documents real-life use of the licensed drug. The pre-approval clinical trial phase might be shortened to three years, with 40 to 50 percent of all candidate compounds that enter this stage completing it. This prediction model will allow developers to recoup costs and enjoy a longer life cycle under patent protection; it also will require pharma to pay closer attention to newly licensed drugs' real-life applications.
Drug development's approval process will face complication and friction in the future because of increasing globalization that will require it to be introduced in an internationally harmonized fashion. The new clinical trials package should be completed by 2015, but resistance to any deviation from the well-established clinical-development process should be considered during the transition. There is, however, no other alternative—restoring productivity is always a question of survival for the pharmaceutical industry.
Hermann Mucke is founding director of HM Pharma Consultancy. He can be reached at office@hmpharmacon.com. Michael Goodman is general manager of advances reports at Cambridge Healthtech Associates. He can be reached at mgoodman@chacorporate.com.
















FDA Grants Priority Review to Regeneron’s Eylea for Macular Edema Following Retinal Vein Occlusion
April 18th 2025Regulatory action was based on data from the Phase III QUASAR trial, which demonstrated that Eylea HD dosed every eight weeks achieved non-inferior visual acuity outcomes compared to Eylea in patients with macular edema following retinal vein occlusion.

Addressing Disparities in Psoriasis Trials: Takeda's Strategies for Inclusivity in Clinical Research
April 14th 2025LaShell Robinson, Head of Global Feasibility and Trial Equity at Takeda, speaks about the company's strategies to engage patients in underrepresented populations in its phase III psoriasis trials.

New Insights Into T Cell Exhaustion and Inflammation in Long COVID
April 17th 2025Nigel McCracken, chief operating officer, Virax Biolabs, discusses new findings that reveal altered cytokine activity and evidence of T cell exhaustion in long COVID patients, providing deeper insight into post-infection immune disruption.

Key Findings of the NIAGARA and HIMALAYA Trials
November 8th 2024In this episode of the Pharmaceutical Executive podcast, Shubh Goel, head of immuno-oncology, gastrointestinal tumors, US oncology business unit, AstraZeneca, discusses the findings of the NIAGARA trial in bladder cancer and the significance of the five-year overall survival data from the HIMALAYA trial, particularly the long-term efficacy of the STRIDE regimen for unresectable liver cancer.

Amgen’s Imdelltra Demonstrates Significant Overall Survival Improvement in Small Cell Lung Cancer
April 16th 2025In the Phase III DeLLphi-304 trial, patients with small cell lung cancer administered Imdelltra achieved a statistically significant and clinically meaningful improvement in overall survival compared to standard-of-care chemotherapy.


